
Retinitis Pigmentosa
What is Retinitis Pigmentosa?
The retina is a specialized layer of neural tissue that coats the back of the eye and is responsible for helping you to see. Retinitis pigmentosa (RP) is the name given to a group of inherited diseases where there is a genetic defect in the body that causes the retina to slowly stop working.
What are the symptoms of Retinitis Pigmentosa?
The most characteristic symptom is difficulty with night vision, and loss of side vision. Certain patients may have more problems with center vision and difficulty with bright lights. Difficulty with night vision is termed night blindness, often severely limiting function in the dark. As the disease progresses the side or peripheral vision continues to get worse. In the advanced stages of the disease only a small central island of vision may remain, termed tunnel. It is rare for patients to lose all vision in both eyes.
How is Retinitis Pigmentosa diagnosed?
Retinitis Pigmentosa is diagnosed based on a clinical examination and often confirmed with more advanced testing. Your ophthalmologist can diagnose retinitis pigmentosa by dilating the pupils and examining the retina. Often there are clumps of pigment noted at the back of the eye (figure 1). Further testing with formal visual field test can be done to tell how much peripheral vision is remaining. A special test called an electroretinogram (ERG) can test the electrical function of the retina. In some cases the exact genetic defect can be identified through genetic testing. The tests are more likely to give a meaningful answer if a number of affected family members can all be tested at the same time.
How is RP inherited?
Most people with RP inherit the gene mutations from one or both of their parents. A mutated gene causes the nerve cells in the retina to function abnormally. There are currently over 100 different identified genetic defects in many different genes that can cause retinitis pigmentosa. The genetic mutations are usually passed on in one of three different patterns: autosomal dominant, autosomal recessive or X-linked. In autosomal dominant disease typically one parent has RP and passes the disease to 50% of their children. In autosomal recessive disease, neither parent has RP but both have abnormal genes. In this case 25% of their kids will get two bad copies and have RP. Lastly in X-linked RP the mother carries the mutation and usually does not have RP. 50% of her sons will have RP and none of her daughters will be affected.
Is there any treatment for Retinitis Pigmentosa?
Currently there is little available, but there is hope that new therapies will help in the near future. Since RP is caused by a genetic defect, a correction of the defect is really needed in order to stop the disease. So-called gene therapy is currently undergoing limited testing throughout the world but is currently unavailable outside of the limited clinical trials. There is controversial data indicating that high dose vitamin A (retinyl palmitate, 15,000 IU) may slow down the disease. Such large doses of vitamin A may be harmful if a patient ends up taking the medication for potentially decades. Supplements of vitamin E alone may make RP worse. Vitamin E will not be harmful if taken with Vitamin A or in the presence of a normal diet. It is important to seek the advice of your ophthalmologist regarding safe vitamin use.
What is the prognosis of Retinitis Pigmentosa?
RP is usually diagnosed in adolescents and young adults. Unfortunately it is a progressive disorder. The rate of vision loss varies from person to person. Most people continue to have some useful vision for their entire life. The best predictor of the amount and rate of vision loss is the history of affected family members. If other affected family members with RP have kept useful vision over their life, then typically the affected person will have useful vision for their life. Since people with RP can also develop other eye disease such as cataracts it is important to see an ophthalmologist every year.
Where can I find more information?
The Foundation Fighting Blindness
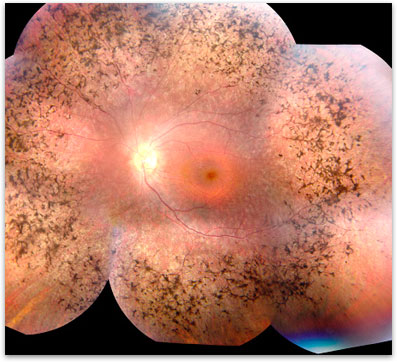
Figure 1. Classic RP showing the pigmentary clumping under the retina.